
Introduction
The Loomis-Wood for Windows program package (abbreviated further to LWW program) is designed for assigning vibration-rotation spectra from Fourier-transform (FT) infrared (IR) spectra. It exists currently in three versions for:
- Symmetric top molecules (program LWW.exe)
- Asymmetric top molecules (program LWWa.exe)
- Molecules with large amplitude motions (program LWWl.exe)
It integrates several approaches used previously by computer programs of this type, giving in separate windows of this application program simultaneous access to:
- The plot of the IR spectrum with labels of predicted/assigned lines in the Spectrum window. The plot of the spectrum is directly linked with its peaklist (table of peak wavenumbers and intensities).
- Listings of wavenumbers in individual spectral branches ( Assignments window ) in which the reproduction of
the IR spectrum (
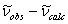 ) is displayed and where assignments can be marked
interactively. Assigned transitions are marked correspondingly in the
Spectrum window.
) is displayed and where assignments can be marked
interactively. Assigned transitions are marked correspondingly in the
Spectrum window. - Symbolic representation of sequences of transitions in Loomis-Wood (LW) diagrams, which translates the numerical information from the wavenumber listings of branches into a graphical representation where visually recognizable patterns provide a powerful tool for control of the assignment. This graphical (LW) representation of the IR spectrum can be also used for making assignments.
- The program allows simultaneous displaying of several LW diagrams that are mutually linked by lower state combination difference (LSCD) relations. This link makes the series of transitions terminating in common upper levels appear as visually recognizable patterns of the same shape. The quantitative measure of similarity of continuous patterns in LW diagrams is displayed together with the interactive control of these diagrams. This is of great help especially in assigning spectra with resonances and/or high density of lines.
The flexibility and effectiveness of the assignment procedures is based on the following assumptions:
- The lower state of the band(s) being assigned (not necessarily the vibrational ground state) is known with accuracy allowing LSCD checking.
- All rovibrational energies are internally stored in a table of energies (labeled with appropriate quantum numbers). This is obviously necessary for LSCD checking of assignments when the lower level energies cannot be expressed in a closed polynomial form (i.e. hot bands of symmetric top molecules with the lower state being a degenerate level of E symmetry, all cases of asymmetric top bands, etc.).
- The program has implemented a simple and flexible calculation of a correction function to upper state energies, which makes possible a quasi-automated assignment procedure (assigning whole series of transitions by ‘just one mouse-click‘) once the correctness of assignments has been verified by LSCD checking.
- The assigned data can be exported to files, to be used in fitting with more complex Hamiltonian models and the refined energy levels imported back into the assignment program. This option can be in the future incorporated directly into the current program.
- Rovibrational energies, refined by least-squares fitting in an external program, can be imported into the LWW program and replace the previously used energies. This provides a possibility of iterative improvement of energies of the upper vibrational state and significant acceleration of the assignment procedure.